


")






21 Abr Características radiológicas en GA1: revisión Radiopaedia marzo 2023
Revisión de Radiopaedia de marzo de 2023 sobre las características radiológicas de la GA1, con la presentación de 6 casos junto a las imágenes neurorradiológicas.
Presentación clínica
La presentación es variable:
- Macrocefalia en los primeros meses de vida
- Asintomáticos.
- Encefalopatía aguda después de una infección o un estado catabólico agudo
- Presentación inicial puede parecerse a la encefalitis viral o la encefalomielitis diseminada aguda ( ADEM ).
- Síntomas extrapiramidales con afectación del estriado y necrosis .
- Algunos inicio insidioso y otros encefalopatía progresiva o son asintomáticos.
Patología
- Causada por la deficiencia hereditaria de la enzima glutaril-CoA deshidrogenasa (GCDH) que conduce a la acumulación de ácido glutárico y ácido 3-hidroxiglutárico en el cerebro y los fluidos corporales, incluida la orina (de ahí el nombre de aciduria glutárica). La aciduria glutárica tipo II es una enfermedad diferente causada por deficiencias enzimáticas no relacionadas .
Marcadores
Los análisis de sangre, orina y LCR de rutina para los metabolitos mencionados anteriormente pueden ser engañosos, ya que la excreción es solo intermitente incluso durante episodios de descompensación aguda. Por lo tanto, la imagen tiene un papel importante que desempeñar en estas situaciones.
Genética
El diagnóstico definitivo de aciduria glutárica tipo 1 se puede establecer mediante un análisis basado en el ADN, en busca de mutaciones en el gen GCDH en el cromosoma 19
Características radiográficas
La resonancia magnética es la modalidad de elección en la evaluación de la aciduria glutárica tipo 1.
-
- En niños gravemente afectados se observan
- Anomalías de los ganglios basales bilaterales con inflamación inicial que posteriormente progresa a atrofia y necrosis.
- Pueden verse afectadas la sustancia negra y los núcleos dentados.
- Hiperintensidad de los tractos tegmentales a lo largo del piso del cuarto ventrículo.
- La mielinización retardada es otro hallazgo en los lactantes gravemente afectados
- Las características comunes
- Macrocefalia
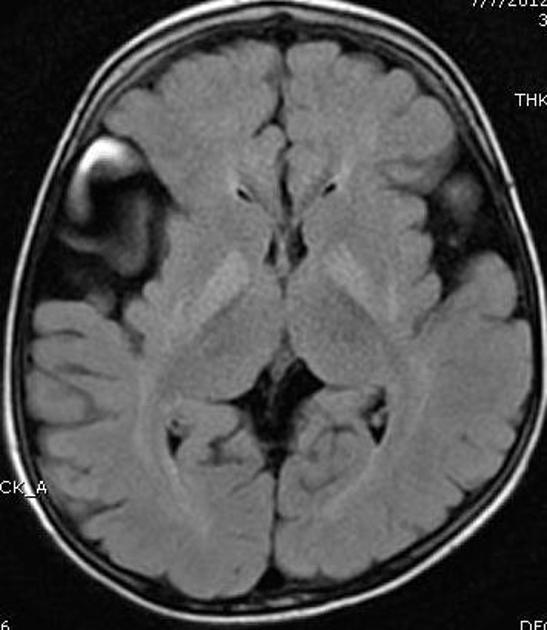
- Expansión de los espacios subaracnoideos y del LCR anteriores a los polos temporales y en las fisuras de Silvio. Sigue sin resolverse si estos amplios espacios del LCR representan quistes aracnoideos en lugar de atrofia y subopercularización de las fisuras de Silvio.
- En niños gravemente afectados se observan
Los hallazgos descritos anteriormente no son específicos de forma aislada, sino una combinación de ellos en un niño macrocefálico con síntomas extrapiramidales altamente sugestivos, si no patognomónicos .
Características de la señal:
- T1: señal baja
- T2/FLAIR: señal alta
- GE/SWI: sin efecto de susceptibilidad
- DWI: difusión restringida de forma aguda
- T1 C+ (Gd): sin realce
- Espectroscopia de RM: pico de lactato dentro de los ganglios basales de forma aguda
Con la expansión de los espacios subaracnoideos de convexidad, las venas puente que discurren son susceptibles de romperse con traumatismos menores, y estos pacientes pueden presentar hemorragias subdurales. En este contexto, el radiólogo debe estar familiarizado con los hallazgos imagenológicos de la aciduria glutárica tipo I para no realizar un diagnóstico erróneo de lesión no accidental
Tratamiento y pronóstico
Se debe buscar un diagnóstico posnatal temprano ya que un tratamiento temprano antes de la descompensación metabólica tiene la mejor oportunidad de prevenir el deterioro neurológico (una vez que ocurre una crisis metabólica, la afectación de los ganglios basales es inevitable). Por esta razón, todos los hermanos de un niño afectado y todos los embarazos futuros deben someterse a pruebas de detección de la enfermedad. Sin embargo, algunos casos son progresivos a pesar de todo el tratamiento adecuado .
El tratamiento en la etapa aguda toma la forma de prevención y corrección del estado catabólico . En la etapa crónica, se debe dar una dieta baja en proteínas con suplementos de carnitina y riboflavina.
Diagnóstico diferencial
- macrocefalia : algunas otras leucodistrofias se presentan con macrocefalia 8 ; estos incluyen la enfermedad de Alexander , la enfermedad de Canavan , la aciduria L-2-hidroxiglutárica y la leucoencefalopatía megalencefálica con quistes subcorticales.
- causas de ganglios basales bilaterales de alta intensidad de T2: aminoacidopatías, enfermedades mitocondriales, enfermedad de Wilson , síndrome de Zellweger
- expansión benigna de los espacios subaracnoideos (BESS): aunque los espacios de convexidad prominentes son características comunes, BESS no se asocia con espacios de LCR prominentes en las fisuras de Silvio o los polos temporales anteriores. Tampoco se aprecian cambios parenquimatosos.
- lesión no accidental (NAI): la aciduria glutárica tipo 1 es un diferencial importante para NAI, ya que puede presentarse con hematomas subdurales