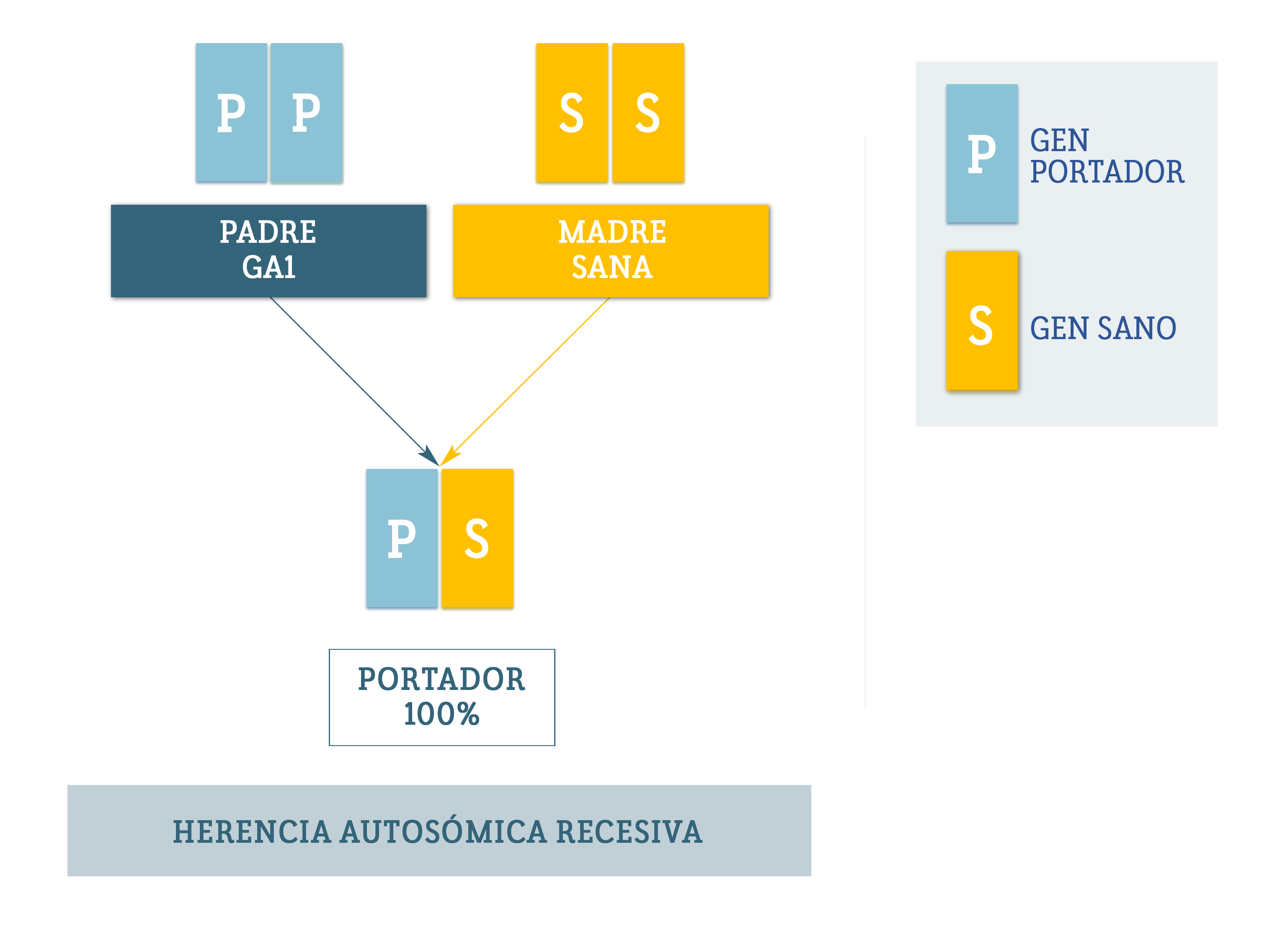
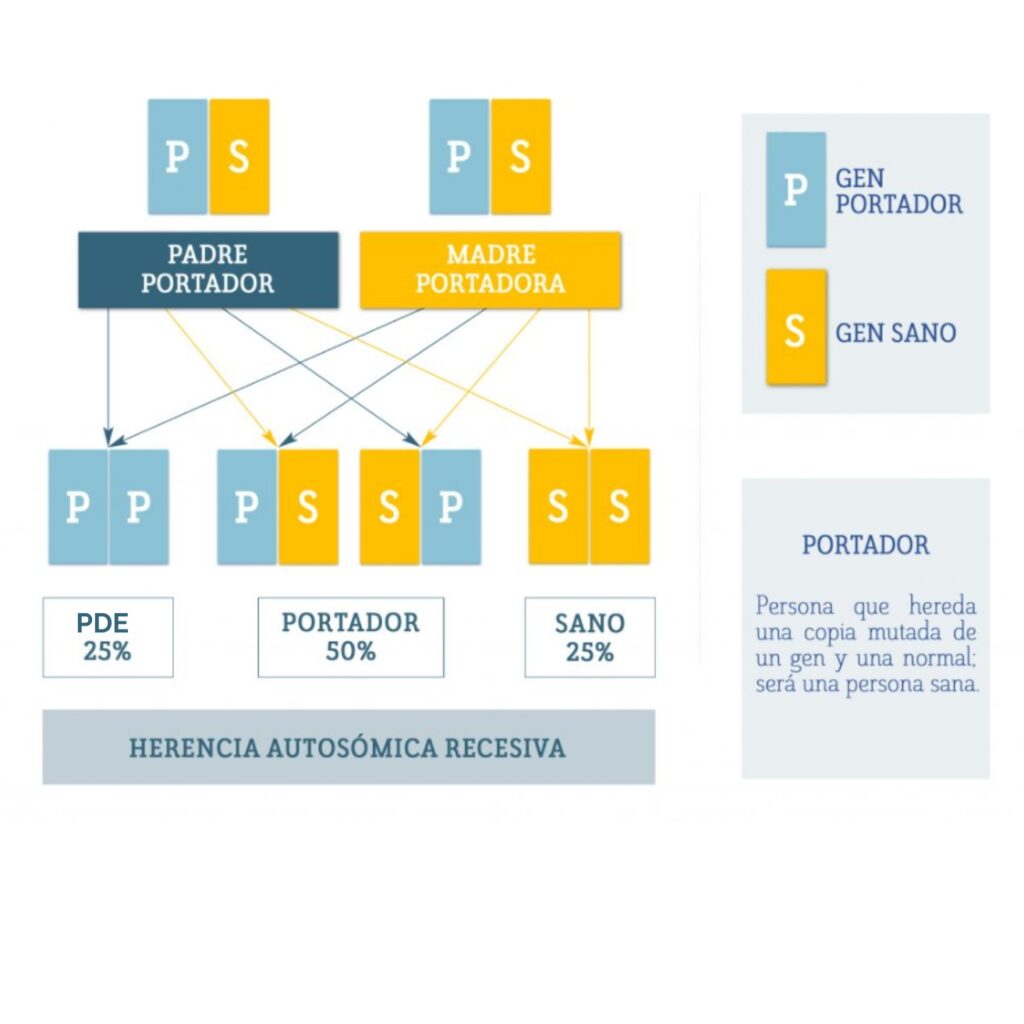
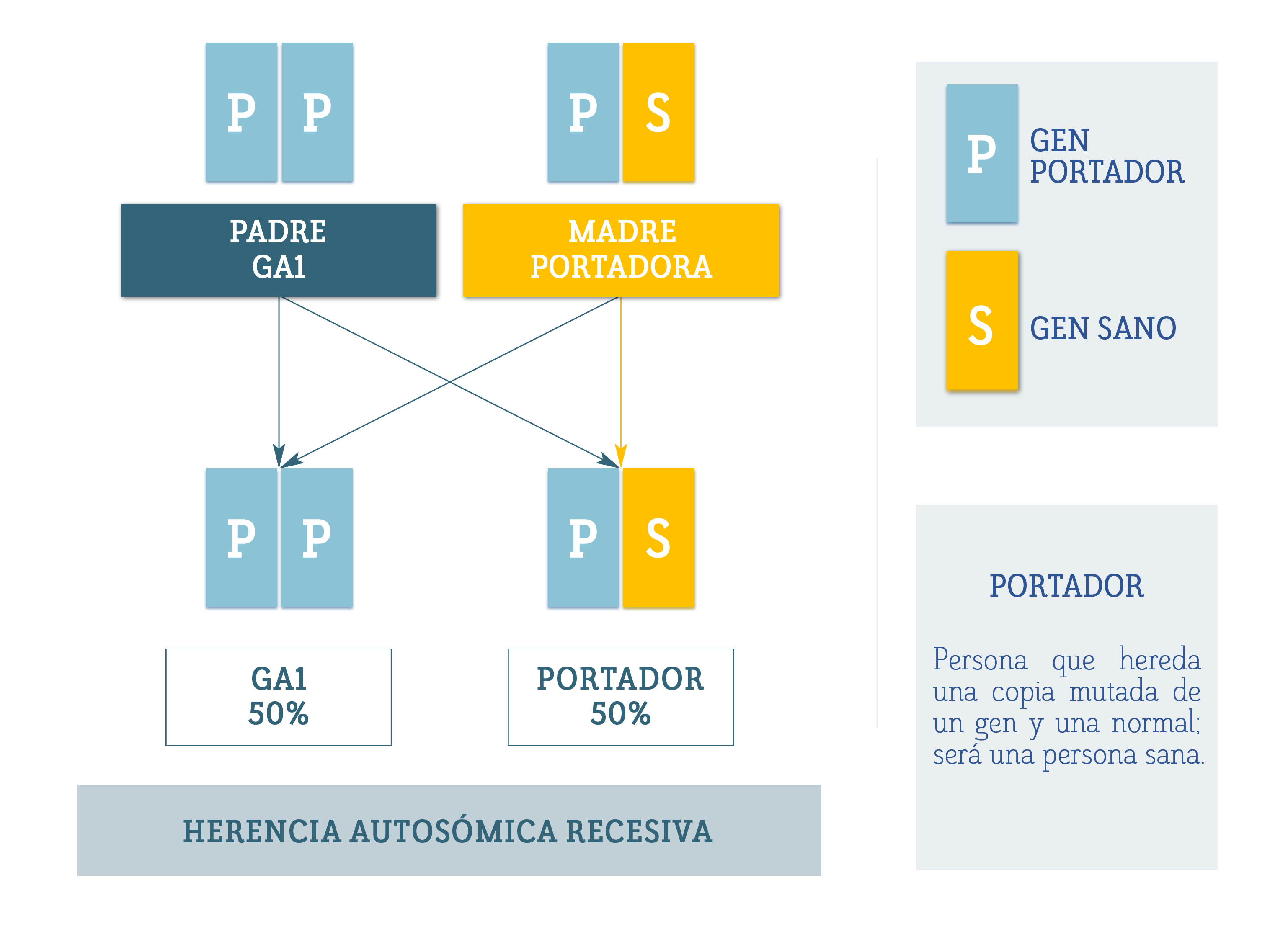
La epilepsia dependiente de piridoxina (PDE) es una enfermedad causada por alteraciones en el gen ALDH7A1, ubicado en el cromosoma 5. Este gen es responsable de producir una enzima llamada antiquitina, que desempeña un papel clave en el cerebro.
La antiquitina participa en la descomposición de la lisina, un aminoácido esencial. Actúa en una ruta metabólica específica, conocida como la vía de degradación de la lisina, en la que su función es facilitar la eliminación de ciertos compuestos. Cuando esta enzima no funciona correctamente, se acumulan sustancias neurotóxicas como AASA, P6C y ácido pipecólico, que afectan el sistema nervioso y provocan las crisis epilépticas típicas de la enfermedad.
«La enzima α-AASA deshidrogenasa oxida α-AASA a ácido α-aminoadípico; una deficiencia de esta enzima resulta en la acumulación de ácido pipecólico,12–14 α-AASA y el Δ1 -piperideína-6-carboxilato (Δ1 -P6C). Se postula que el Δ1 -P6C acumulado se une al vitámero activo de la piridoxina (piridoxal 5′-fosfato) a través de una condensación de Knoevenagel, y se utilizan dosis farmacológicas de piridoxina para superar la deficiencia secundaria de piridoxal 5′-fosfato. La α-AASA deshidrogenasa está codificada por el gen ALDH7A1 (NM_001182), que también se conoce como antiquitina. ALDH7A1 se encuentra en el cromosoma 5q32.2, que contiene una transcripción de 4.964 pares de bases y 539 aminoácidos divididos entre 18 exones. Hasta el momento, se han publicado más de 165 variantes patógenas en ALDH7A1» *2
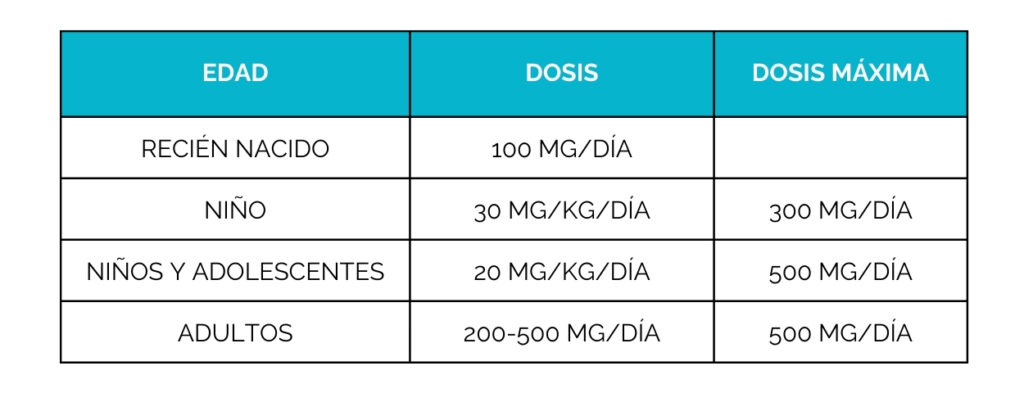
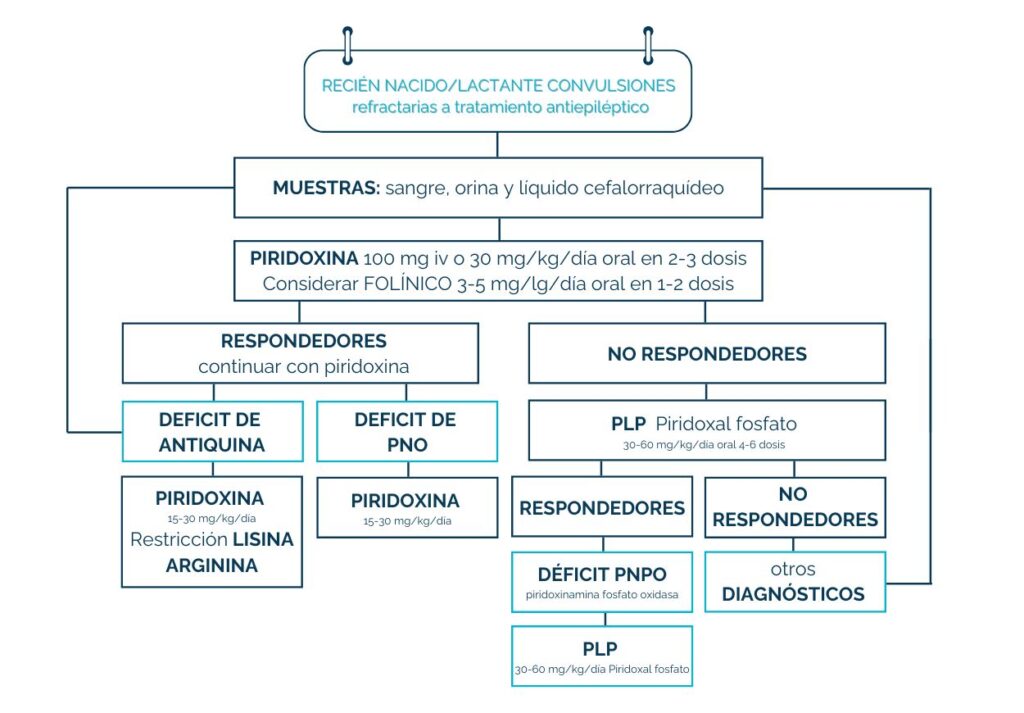
Dado que el problema subyacente está relacionado con la lisin, los fármacos tradicionales para la epilepsia no resultan eficaces en estos pacientes. Sin embargo, el tratamiento con vitamina B6 (piridoxina) permite compensar parcialmente este defecto y ayuda al cerebro a funcionar mejor, evitando las convulsiones.A pesar de ello, la toxicidad neuronal persiste en algunos casos, lo que ha llevado al desarrollo de terapias complementarias dirigidas a reducir los niveles de lisina en el organismo.
Estas estrategias incluyen:
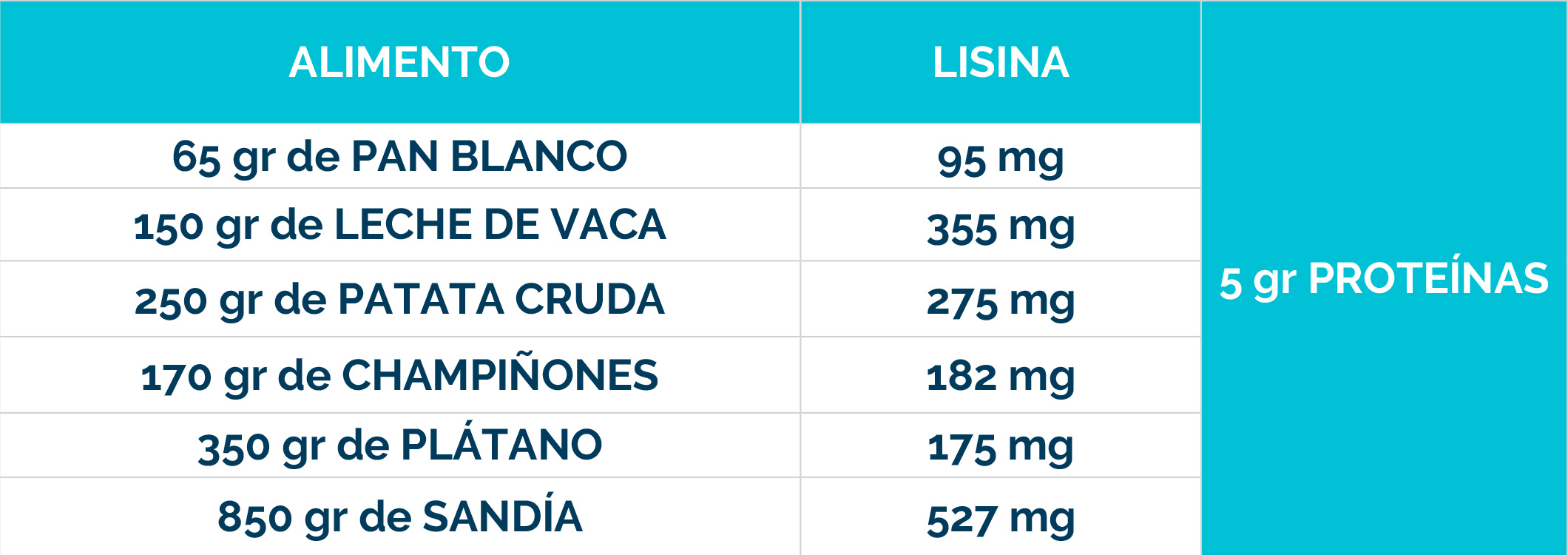
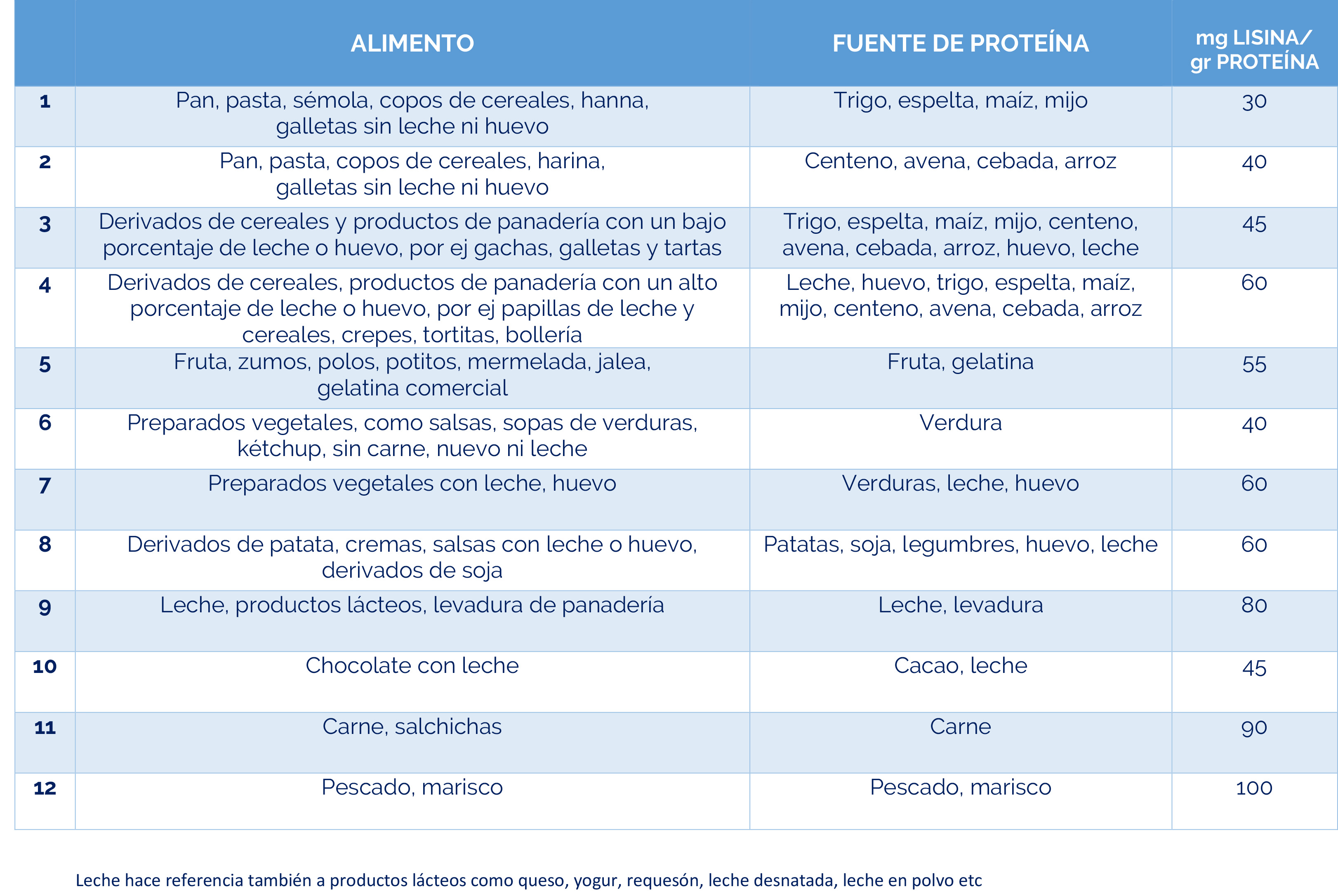
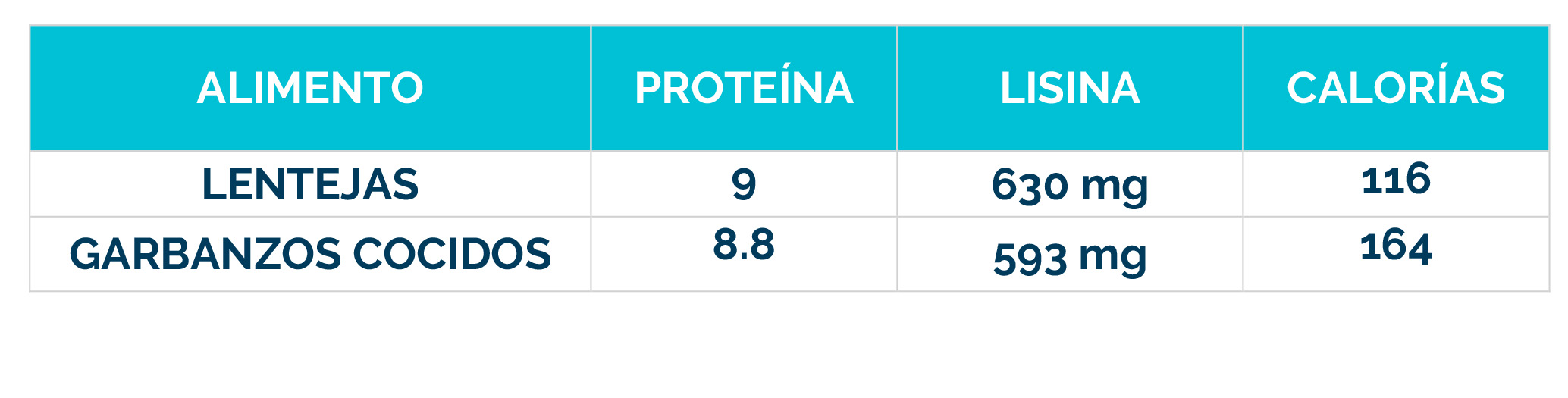
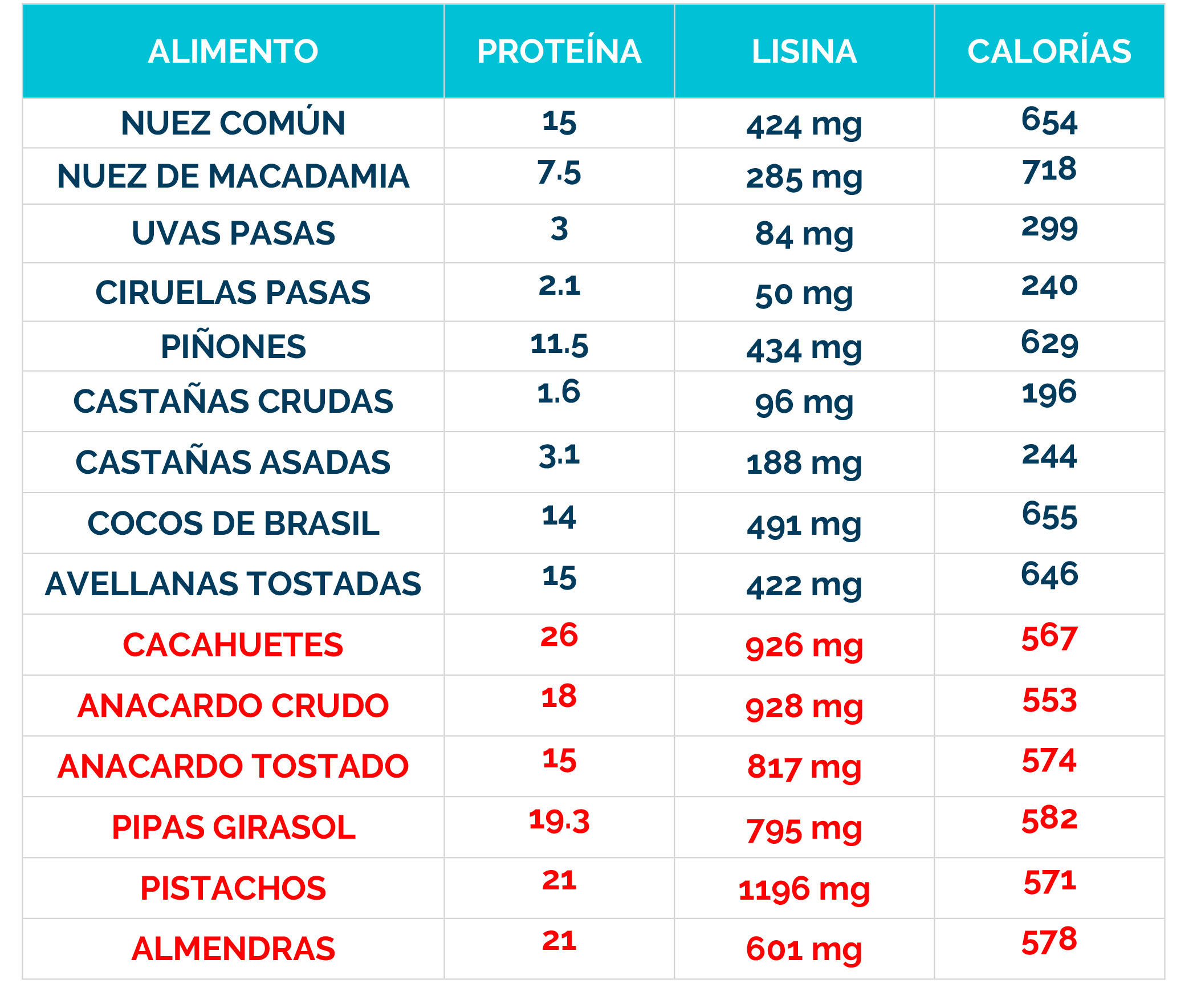
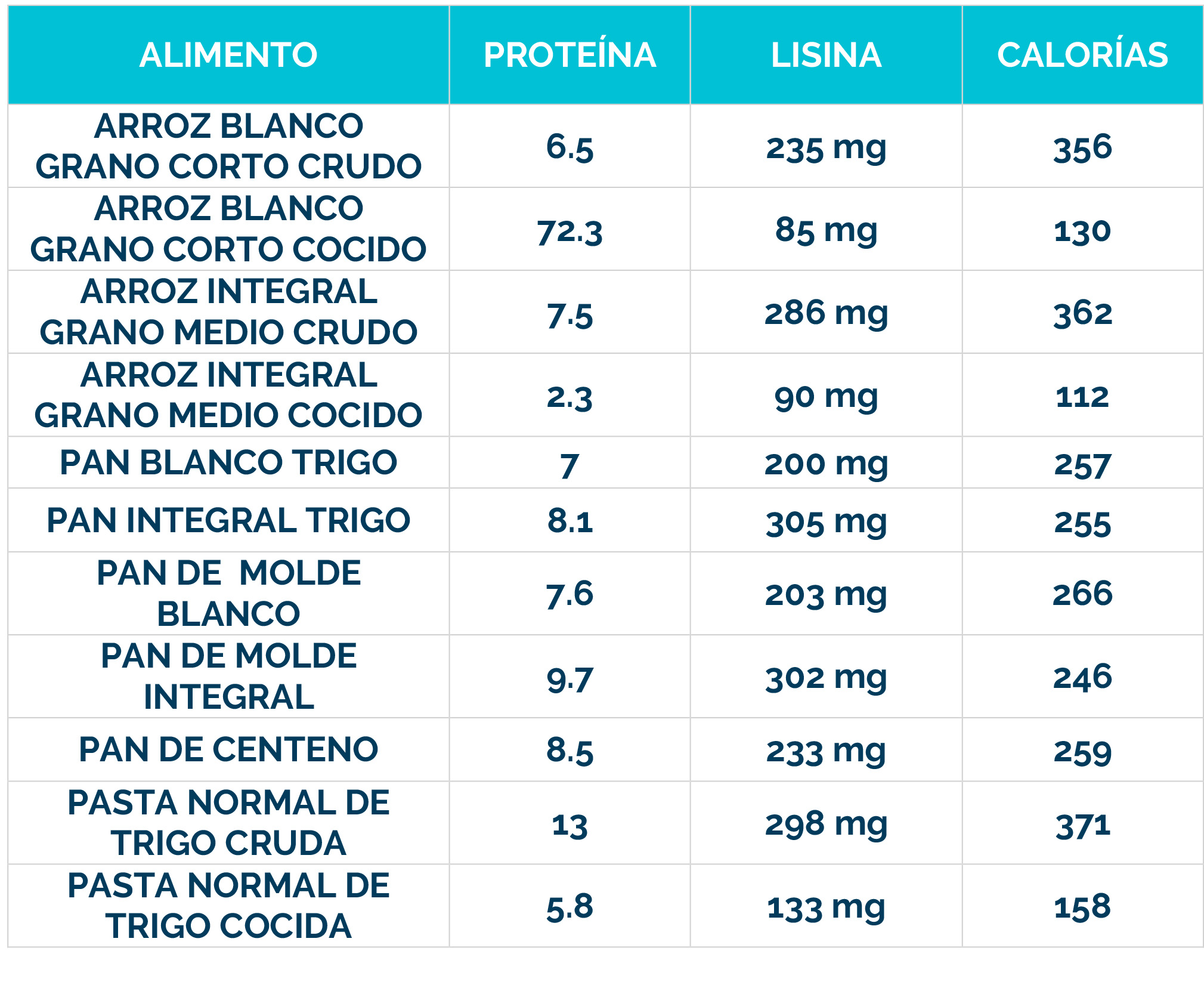
– Dieta restringida en lisina, que reduce la cantidad de este aminoácido en el cuerpo.
– Suplementos de arginina, que compiten con la lisina por el transporte al cerebro, limitando su acceso al sistema nervioso.
La combinación de dieta baja en lisina, suplementos de arginina y vitamina B6 se conoce como «Triple Terapia». Se ha demostrado que este enfoque es seguro y bien tolerado, además de reducir los biomarcadores neurotóxicos y mejorar el control de las convulsiones, el desarrollo psicomotor y el comportamiento.
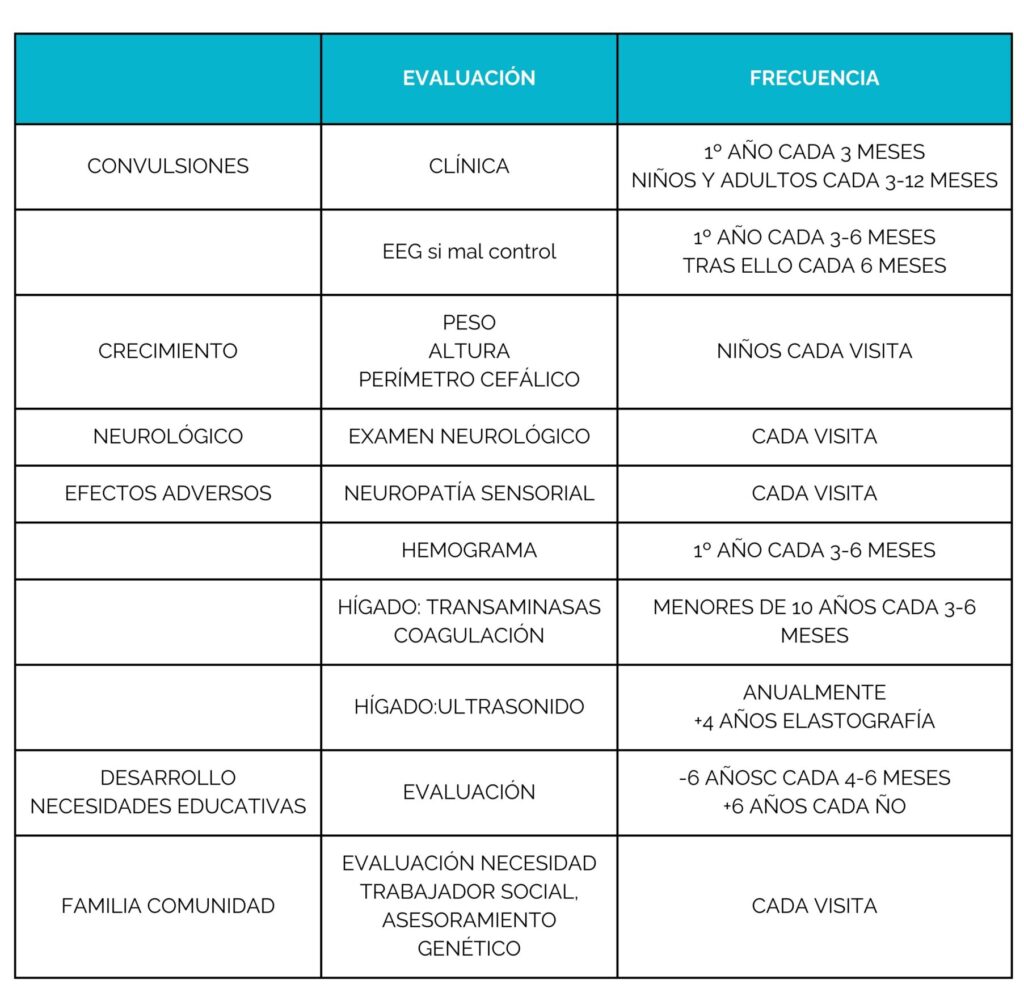
Investigar la causa genética de la PDE es clave para lograr un diagnóstico más temprano y seguir avanzando en la búsqueda de tratamientos más efectivos en el futuro.



")